
Вход на сайт
Новости
XMD перевод документации
пятница, марта 24, 2017 - 10:01
Содержимое:
Первый вариант перевода здесь. Пока не причесано, но работа продолжается...
Недокументированные возможности Android
пятница, марта 10, 2017 - 09:05
Содержимое:
10 возможностей Android, о которых вы могли не знать https://hi-tech.mail.ru/news/android-features/
Вход на сайт - через соцсети!
среда, марта 1, 2017 - 17:31
Содержимое:
Запущен модуль авторизации пользователей через социальные сети - OneAll Social Login. Пока вход через ВКонтакте и mail.ru
Страницы
Пакет программ XMD. Динамические опыты.
Статья опубликована: журнал "Системный администратор", номер №10, 2014г. под названием:
Пакет программ XMD
Физические эксперименты за компьютером.
Перевод документации к программе XMD на русский язык можно найти здесь - http://learn2prog.ru/xmd-doc-translate
Продолжаем знакомство с программами молекулярной динамики (МД). В [1] мы познакомились с программным пакетом XMD, установили его на компьютер и попытались сделать кристаллическую решетку алмаза. Также, в [1] говорилось, что для постановки динамических экспериментов в МД к описанию кристаллической решетки в программном файле необходимо подключить файлы потенциалов, где учитываются силы взаимодействия между атомами. Понятно, что динамические опыты и есть цель создания подобных программных пакетов. Поэтому покажем как использовать для этого XMD, применяя знания физики на уровне старших классов средней школы, для несложных, но достаточно интересных, физических опытов с металлами.
Применение файлов потенциалов
Файл потенциалов представляет собой обычный текстовый файл, в котором значения потенциальной энергии взаимодействия частиц записываются по определенным правилам. Для метода вложенных атомов (EAM - embedded atom method), с которым мы также познакомились в [1], эти правила описаны в справке (загляните в папку /doc программного каталога XMD или на сайт проекта). Ряды значений потенциальной энергии записываются в энергетических единицах определяемых командой eunit и располагаются после идентификации функции взаимодействия, например строка
POTENTIAL PAIR 1 1 2000 1.2 6.3
определяет потенциал парного взаимодействия между атомами типа 1. Ряды значений функции (таблица значений) будут содержать 2000 чисел, далее следуют два параметра, которые определяют входной диапазон для таблицы. В примере для парного потенциала, это входные расстояния, которые принимают значения от 1.2 до 6.3 ангстремов.
В папке /examples программного каталога XMD можно найти готовые файлы для золота, кобальта, а также соединений никель-алюминий, ниобий-алюминий, рутений-алюминий. Проверенные файлы потенциалов для других простых веществ и соединений можно найти в сети Интернет. например в [2], [3].
Мы будем использовать для наших опытов файл потенциалов железа, одного из самых распространенных металлов, составленный Дианой Фаркаш, который можно скопировать с сайта проекта XMD [2]. Рассмотрим подробнее этот файл, открыв его в текстовом редакторе (см. Листинг 1).
Листинг 1. Выдержки из файла fefarkas.txt
eunit K
calc A0=2.87
calc MassFe=55.85
calc DTIME=1e-16
dtime DTIME
POTENTIAL SET EAM 1
POTENTIAL PAIR 1 1 3000 9.920413E-01 4.095403E+00
8.935990E+04 8.920155E+04 8.904339E+04 8.888540E+04
8.872760E+04 8.856996E+04 8.841251E+04 8.825523E+04
8.809813E+04 8.794121E+04 8.778446E+04 8.762789E+04
...
POTENTIAL DENS 1 3000 9.920410E-01 4.095400E+00
1.641865E+00 1.636743E+00 1.631639E+00
1.626552E+00 1.621483E+00 1.616432E+00 1.611398E+00
1.606382E+00 1.601382E+00 1.596400E+00 1.591436E+00
...
POTENTIAL EMBED 1 3000 3.398751E-02 3.315712E+00
-1.974536E+04 -1.994018E+04 -2.012917E+04 -2.031256E+04
-2.049056E+04 -2.066335E+04 -2.083112E+04 -2.099404E+04
-2.115229E+04 -2.130600E+04 -2.145533E+04 -2.160042E+04
...
calc A0=2.87
calc MassFe=55.85
calc DTIME=1e-16
dtime DTIME
POTENTIAL SET EAM 1
POTENTIAL PAIR 1 1 3000 9.920413E-01 4.095403E+00
8.935990E+04 8.920155E+04 8.904339E+04 8.888540E+04
8.872760E+04 8.856996E+04 8.841251E+04 8.825523E+04
8.809813E+04 8.794121E+04 8.778446E+04 8.762789E+04
...
POTENTIAL DENS 1 3000 9.920410E-01 4.095400E+00
1.641865E+00 1.636743E+00 1.631639E+00
1.626552E+00 1.621483E+00 1.616432E+00 1.611398E+00
1.606382E+00 1.601382E+00 1.596400E+00 1.591436E+00
...
POTENTIAL EMBED 1 3000 3.398751E-02 3.315712E+00
-1.974536E+04 -1.994018E+04 -2.012917E+04 -2.031256E+04
-2.049056E+04 -2.066335E+04 -2.083112E+04 -2.099404E+04
-2.115229E+04 -2.130600E+04 -2.145533E+04 -2.160042E+04
...
Посмотрев этот листинг, можно догадаться, что файл потенциалов — это такой же командный файл XMD. Приведем некоторые пояснения к использованным в этом файле командам. Как уже говорилось, eunit устанавливает энергетические единицы для потенциальных сил, в данном случае это - Кельвины. Далее, с помощью команды calc создаются переменные определяющие размер элементарной ячейки железа - A0, массу атома железа – MassFe, а также определяется временной шаг моделирования для молекулярной динамики с помощью команды dtime.
Команда calc в XMD обеспечивает внутренний язык для вычислений. Например, если задано
CALC x = 2^(1/3)
то это означает, что создается переменная x, которая равна 2 в степени 1/3. И после этого уже можно использовать переменную x различных командах.
Далее, строка файла
POTENTIAL SET EAM 1
определяет, что будет использоваться метод вложенных атомов - EAM. Ниже записываются функции для парного взаимодействия PAIR и электронной плотности DENS атомов железа, которые зависят от расстояния между атомами, а также комбинированная «функция вложения» EMBED, зависящая уже от электронной плотности.
Отметим, что разделителями значений для этих функций в файле является пробел, табуляция или перенос строки, и этим обеспечивается достаточная гибкость ввода информации.
Нужно сказать, что файлы потенциалов тщательно проверяются исследователями на соответствие реальным физическим свойствам материалов, таким образом постоянно повышается достоверность симуляций с помощь метода МД. А для самого исследователя создание своего файла потенциалов является предметом признания его вклада в науку.
Составление XMD-программы для динамической симуляции
Теперь постараемся показать на примере, как в XMD осуществляются динамические симуляции. Наша задача - составить командный файл, в котором будут описаны условия для нашего динамического опыта с железом, а опыт будет следующим. С помощью XMD создадим кристаллит нужного размера, подключим к программе файл потенциалов, установим физические свойства среды и окажем давление на часть нашего кристаллита с помощью команды extforce. Наша xmd-программа будет имитировать воздействие на кристаллическую решетку «извне» а мы будем наблюдать за движениями атомов с помощью программы визуализации xmdview и запишем некоторые физические параметры выбранных частиц в текстовый файл.
Чтобы построить кристаллит, составленный из атомов железа необходимо совсем немного углубиться в физику. Состояние железа, в котором оно находится при температуре до 769 °С носит название «альфа-железо». Кристаллическая решетка железа в таком состоянии относится к объемно-центрированной кубической (ОЦК) группе. В элементарной ячейке при таком строении вещества атомы железа располагаются в пространстве в углах куба со стороной равной постоянной решетки (размер элементарной ячейки) и в его центре. Построить такую решетку в XMD достаточно просто
#Чтение файла потенциалов
read fefarkas.txt
#построение ОЦК решетки железа
box 5 5 5
fill particle 2
1 0 0 0
1 1/2 1/2 1/2
fill go
#постоянная решетки
scale A0
read fefarkas.txt
#построение ОЦК решетки железа
box 5 5 5
fill particle 2
1 0 0 0
1 1/2 1/2 1/2
fill go
#постоянная решетки
scale A0
в этом фрагменте XMD-программы мы создаем кристаллит состоящий из 5*5*5 элементарных ячеек кристаллической решетки железа с помощью box и fill, а в команде scale A0 используется переменная A0, уже определенная нами ранее в файле потенциалов, соответствующая постоянной решетки альфа-железа. Файл потенциалов подключается с помощью команды read fefarkas.txt.
Отметим, что со значка решетки «#» в XMD начинается комментарии, которыми можно снабдить код для объяснения того что происходит в программе.
Следующим шагом необходимо назначить массы атомам железа. Для этого нам необходимо познакомиться еще с одной важной командой из арсенала XMD — select. Команда select — выбирает атомы в кристаллите по определенному условию, для осуществления с ними каких-либо действий. Например, для назначения массы всем атомам нашего кристаллита достаточно следующего кода
select all
mass MassFe
mass MassFe
при этом используется переменная MassFe, которая была определена ранее в файле потенциалов fefarkas.txt.
А вот другой вариант кода, с помощью которого выбираются атомы, попадающие в сферу с центром в определенном месте нашего кристаллита, с координатами x = 14, y = 0 и z = 14, выраженными в ангстремах, что соответствует одному из углов нашего кристаллита, и радиусом 5 ангстрем. Целью такого выделения, в нашем случае, является приложение внешней силы к выделенным атомам с помощью команды extforce
select ellipse 14 0 14 5 5 5
extforce 0 0.2e-3 0
extforce 0 0.2e-3 0
сила здесь прикладывается в направлении оси y.
Следующие команды определяют физические свойства среды, а именно температуру 800 Кельвинов (около 530°С) и назначают случайные скорости для всех частиц, соответствующие распределению Максвелла-Больцмана для указанной температуры (clamp)
itemp 800
clamp 800
clamp 800
Отдельно нужно сказать о реализации циклов в XMD. repeat - простая команда цикла, которая выглядит так,
repeat 10
команда 1
команда 2
end
команда 1
команда 2
end
Все команды между repeat и end в этом примере будут повторены 10 раз.
Циклы в XMD используются обычно для команды cmd, которая рассчитывает новые положения частиц через временной шаг, установленный в dtime. В нашем примере с помощью цикла будет осуществляться еще запись необходимых физических параметров на каждом шаге моделирования в текстовый файл с помощью команды write.
Теперь нам осталось привести весь командный файл целиком, что будет сделано в Листинге 2 и дать последние объяснения (обратите внимание также на комментарии в Листинге 2)
Листинг 2. Файл XMD-программы afe_piece.xm
#Чтение файла потенциалов
read fefarkas.txt
#построение ОЦК решетки железа
box 5 5 5
fill particle 2
1 0 0 0
1 1/2 1/2 1/2
fill go
# постоянная решетки
scale A0
# установим массы частиц
select all
# MassFe определена в файле fefarkas.txt
mass MassFe
# установим случайные скорости для атомов, соответствующие температуре 800K
clamp 800
itemp 800
# создаем .cor файл, записываем начальные координаты частиц
write cor fewithextforce.cor
# выделение области для применения внешнего воздействия
select ellipse 14 0 14 5 5 5
# на выбранные частицы действует внешняя сила
extforce 0 0.2e-3 0
# выбираем частицу с номером 210
select index 210
# запись выбранных параметров в начальный момент в файл 210.temp
write file 210.temp sel var i tm ek ep
# цикл из 6000 (600*10) шагов моделирования
repeat 600
cmd 10
write file +210.temp sel var i tm ek ep vx
write cor +fewithextforce.cor
end
read fefarkas.txt
#построение ОЦК решетки железа
box 5 5 5
fill particle 2
1 0 0 0
1 1/2 1/2 1/2
fill go
# постоянная решетки
scale A0
# установим массы частиц
select all
# MassFe определена в файле fefarkas.txt
mass MassFe
# установим случайные скорости для атомов, соответствующие температуре 800K
clamp 800
itemp 800
# создаем .cor файл, записываем начальные координаты частиц
write cor fewithextforce.cor
# выделение области для применения внешнего воздействия
select ellipse 14 0 14 5 5 5
# на выбранные частицы действует внешняя сила
extforce 0 0.2e-3 0
# выбираем частицу с номером 210
select index 210
# запись выбранных параметров в начальный момент в файл 210.temp
write file 210.temp sel var i tm ek ep
# цикл из 6000 (600*10) шагов моделирования
repeat 600
cmd 10
write file +210.temp sel var i tm ek ep vx
write cor +fewithextforce.cor
end
В нашем файле появились новые команды
select index 210
выбирает частицу с номером 210, за параметрами которой мы будем следить и записывать их в файл.
write cor fewithextforce.cor
создает файл с координатами частиц для просмотра движения частиц в программе xmdview, а
write cor +fewithextforce.cor
дописывает, с помощью опции «+», координаты атомов через каждые 10 шагов моделирования в цикле repeat.
Команда
write file 210.temp sel var i tm ek ep
записывает в текстовый файл 210.temp номер, мгновенную температуру, кинетическую и потенциальную энергию частицы, которая выделена с помощью
select index
Осталось произвести запуск моделирования, что выполняется уже рассмотренной нами командой
xmd afe_piece.xm
посмотреть движение частиц можно с помощью
xmdview fewithextforce.cor
Простейшие действия с программой xmdview мы уже обсуждали в [1]. На рис.1 выделены кнопки, с помощью которых можно перейти к следующему/предыдущему состоянию кристаллита, сохраненному в .cor файле
Положение частиц вначале и на 4540 шаге моделирования посмотрите на рис. 1 (обратите внимание на несоответствие положений частиц на границах кристаллита из-за "граничных условий", о них мы говорили в [1]), а температуру, кинетическую и потенциальную энергию и скорость по оси x выбранной частицы на каждом 10 шаге моделирования можно увидеть просмотрев файл 210.temp любым текстовым редактором. Из анализа этих параметров исследователем могут быть сделаны различные выводы.
Листинг 3. Выдержки из файла 210.temp
VAR i tm ek ep vx 1
210 1.116965e+03 1.675447e+03 -4.966887e+04 5.0737e+04
210 1.125387e+03 1.688080e+03 -4.966803e+04 5.0661e+04
210 1.134754e+03 1.702132e+03 -4.966241e+04 5.0553e+04
210 1.145665e+03 1.718497e+03 -4.965199e+04 5.0426e+04
....
210 1.116965e+03 1.675447e+03 -4.966887e+04 5.0737e+04
210 1.125387e+03 1.688080e+03 -4.966803e+04 5.0661e+04
210 1.134754e+03 1.702132e+03 -4.966241e+04 5.0553e+04
210 1.145665e+03 1.718497e+03 -4.965199e+04 5.0426e+04
....
Применительно к нашему опыту, можно проанализировать положение частицы №210 нашего кристаллита. Очевидно, что этот атом после применения внешнего воздействия покидает «свою» элементарную ячейку и проделывает некоторый путь, сталкиваясь при этом с другими частицами и воздействуя на них. Таким образом, мы наблюдаем явление, которое называется в физике диффузией. Рекомендую заинтересовавшимся «поиграть» величиной внешнего воздействия в команде extforce. Таким образом можно определить силу, необходимую для возникновения диффузии.
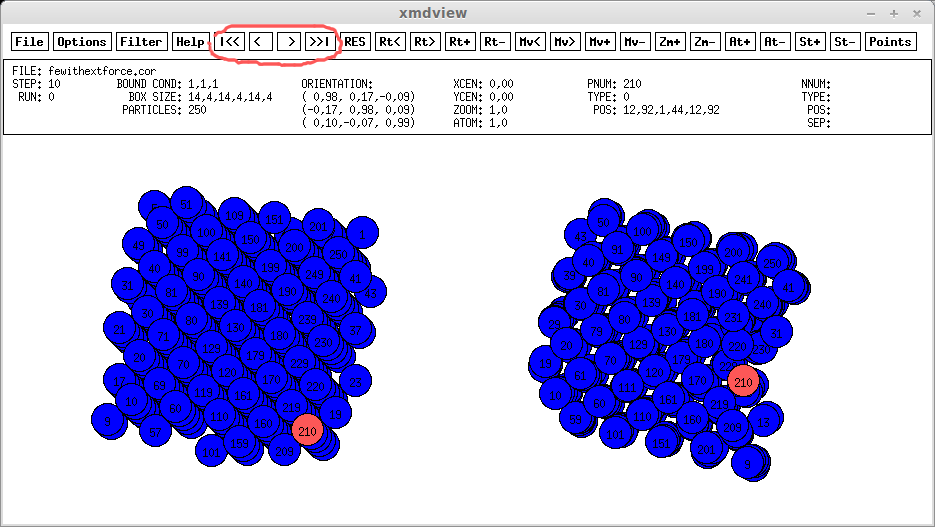
рис.1 Скриншот xmdview на 4540 шаге моделирования (другим цветом выделен атом с номером 210 с помощью прогаммы gimp)
Таким образом, как оказалось, с помощью xmd можно проводить динамические симуляции, позволяющие рассчитывать и наблюдать различные процессы в веществе, сидя в любимом кресле за компьютером и не покидая своей «домашней лаборатории». В следующей статье - об инструментах визуализации XMD.
Литература, ссылки:
1. Системный администратор №7-8(140-141) июль-август 2014, Моделирование «наш» метод познания, с.111-115
2. http://xmd.sourceforge.net/eam.html
3. http://www.ctcms.nist.gov/~cbecker Interatomic Potentials Repository Project
- Войдите или зарегистрируйтесь, чтобы отправлять комментарии
|
Позиция сайта в рейтинге:
|
Дружественные сайты: |