
Запущен проект "Память+"
Читать здесь
О программе молекулярной динамики XMD
На рисунке - программа для просмотра видео vmoviec, один из инструментов "визуализации" пакета XMD.
Статья опубликована: журнал "Системный администратор" в номере №7-8, 2014г. под названием:
Моделирование – “наш” метод познания
(Программа молекулярной динамики XMD).
Перевод документации к программе XMD на русский язык можно найти здесь - http://learn2prog.ru/xmd-doc-translate
Цель данной статьи — знакомство с программным продуктом, созданным для моделирования процессов, происходящих в веществе на уровне атомов, молекул и кристаллических решеток. Речь пойдет о классической программе молекулярной динамики — XMD [1].
Этот материал может быть интересен как учителям информатики, старшеклассникам и студентам (тема «Моделирование» есть в перечне требований к предметным результатам Федерального государственного образовательного стандарта среднего (полного) общего образования), так и инженерам и исследователям различных специальностей (физика, химия, металловедение, материаловедение, дефектоскопия и т.д.). И, наконец, можно с уверенностью сказать, это может быть интересно просто людям любознательным, творческим, которых на просторах интернета не так уж мало.
Компьютерное моделирование – это современный и (в этом сходятся мнения многих исследователей) перспективный метод познания. Компьютерные модели проще и удобнее исследовать в случаях, когда реальные эксперименты затруднены из-за финансовых, физических или других препятствий и описываемый метод моделирования не является исключением.
Программы молекулярной динамики
Молекулярная динамика (МД) или метод молекулярной динамики известен с середины 20 века (первая работа, посвященная МД, вышла в 1957 году – авторы Б. Алдер и Э. Вайнрайт). Изначально разработанный в теоретической физике, этот метод моделирования получил большое распространение в химии и, начиная с 1970-х годов, в биохимии и биофизике. В наше время «нанотехнологий» с помощью средств МД уже изготавливают лекарства и, возможно, в ближайшем будущем возникнут новые специальности, например, «молекулярный инженер-конструктор» [2].
Для описания движения атомов или частиц в этом методе применяется классическая механика, при этом силы межатомного взаимодействия представляются в форме потенциальных сил (см. [3]). Если говорить точнее, в МД рассчитывается движение для каждого отдельного атома. Такой алгоритм осуществляется следующим образом:
- Начальные положения, скорости и массы каждого атома определены;
- Метод использует межатомные потенциалы или, как их еще называют, потенциалы парного взаимодействия (ППВ), которые определяются заранее, как функция потенциальной энергии, зависящая от расстояния между атомами (в простейшем случае);
- Используя ППВ, положения атомов и скорости рассчитывается новое положение частиц через определенный временной интервал , который называется «шаг моделирования». Эти новые положения и скорости становятся исходными для шага вычислений 2, шаги 2, 3, 4 и т. д. повторяются через выбранный временной интервал;
- Как правило, моделирование молекулярной динамики будет включать тысячи таких временных шагов, каждый из которых соответствует доле пикосекунды (10-12 секунды).
Этот алгоритм, по существу, - интеграция ньютоновских уравнений движения материальной точки во времени, решение которых приводит к определенным положениям частиц и скоростям (желающим вникнуть в физику процесса глубже поможет любой поисковик или список литературы приведенный в конце статьи).
В описаниях метода говорится, что МД нельзя противопоставлять натурным экспериментам, однако исследователям для понимания процессов, происходящих в веществе иногда просто необходима визуализация (расчет) движения или расположения атомов и структуры моделируемой системы. В настоящее время возможности персональных компьютеров позволяют проводить моделирование с помощью МД и получать при этом результаты (кстати, очень близкие к результатам настоящих экспериментов), которые еще десять лет назад могли быть получены лишь с помощью суперкомпьютеров. То есть, исследователь при минимальных затратах может получить в свое распоряжение еще один весьма полезный и универсальный инструмент оценки результатов экспериментов [4], [5].
Существует множество программ для моделирования методом МД (список некоторых из них см. в Таблице 1). В основном это иностранные пакеты программирования, для изучения которых потребуется знание английского языка. Хотя, нужно сказать, известны публикации еще советских ученых по этому вопросу - [6], в которых видны основные принципы современных компьютерных систем МД. Однако, массового применения эти разработки так и не получили в связи с известными событиями в нашей стране в 90-х годах прошлого века. Отрадно говорить, что в настоящее время в российской науке возвращается интерес к данной теме (например, вот что нашлось в Google по запросу «российские системы молекулярной динамики» - [5], [7]).
| Название | Сайт поекта | Примечания | Лицензия/Стоимость |
|---|---|---|---|
| CHARMM (Chemistry at HARvard Macromolecular Mechanics) | http://www.charmm.org | Программа МД Гарвардского университета (США). Разработана, в основном, для моделирования экспериментов в биологии. | Для использования в научных учреждениях/$600 |
| LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) | http://lammps.sandia.gov | Одна из самых мощных программ МД. Разработана группой из Сандийских национальных лабораторий (США). Возможность многопроцессорных вычислений при использовании мощностей видеокарт NVIDIA (технология CUDA). | GPL |
| HOOMD (Highly Optimized Object Oriented Molecular Dynamics) | http://codeblue.umich.edu/hoomd-blue/ | Пакет МД Мичиганского университета (США). В программе широко используется язык высокого уровня python. | Использование со ссылкой в публикации на сайт производителя/бесплатная |
| GROMACS (GROningen MAchine for Chemical Simulations) | http://www.gromacs.org/ | Гронингенская Машина для Химического Моделирования - пакет МД, первоначально разработанный группой из Гронингенского университета (Нидерланды). Пакет предназначен, главным образом, для моделирования биомолекул (белки и липиды). | GPL |
| NAMD (NAnoscale Molecular Dynamics) | http://www.ks.uiuc.edu/Research/namd/ | Бесплатная программа для МД, написанная с использованием модели параллельного программирования. Используется для симуляции больших систем (миллионы атомов). Разработана в Иллинойском университете (США). связана с программой визуализации VMD того же университета. | Бесплатная |
Еще один пакет программ, который не вошел в Таблицу 1, но о котором пойдет речь, это — XMD, который представляет собой классическую программу для моделирования методом молекулярной динамики (см. [1]). Предназначен пакет, в основном, для моделирования процессов, происходящих в металлах и керамике. Программа разработана уже достаточно давно в Коннектикутском университете США (англ. University of Connecticut, сокращенно — UConn) под руководством Джона Рифкина (Jon Rifkin), доступна в исходных кодах и распространяется бесплатно под лицензией GPL. Пакет XMD постоянно дорабатывается и на момент написания статьи используется версия программы 2.5.38.
Отличительной особенностью этой программы, для работы с которой используется интерфейс командной строки (Command Line Interface, CLI), является то, что на ее «вход» подается текстовый файл, в котором исследователь с помощью специальных команд описывает необходимую кристаллическую решетку вещества, физические условия проведения эксперимента и контролируемые показатели на выходе. По существу – для работы кроме XMD нужен лишь обычный текстовый редактор (подобный интерфейс еще имеет, например, пакет программ для моделирования LAMMPS). Отсутствие графического интерфейса пользователя имеет даже свои преимущества - дает возможность разработчикам сосредоточиться на отладке численных методов и логике работы программы, а графика появляется лишь там, где она необходима – при визуализации результатов эксперимента (об этом мы обязательно поговорим позднее).
Перевод документации к этой программе на русский язык можно найти здесь - http://learn2prog.ru/xmd-doc-translate
Возможности XMD
Как уже было сказано выше, пакет XMD предназначен, главным образом, для симуляции различных процессов в металлах, их сплавах и керамике. Физические процессы, происходящие в металлах вполне понятны даже школьникам, поэтому XMD – именно та программа, с которой вполне «можно начать» знакомиться с увлекательным миром компьютерного моделирования.
Представляем читателю, совсем «немаленький» список опций этой программы (в скобках указаны команды, используемые во входном файле):
- использование одного или нескольких командных файлов, при этом встроенный макропроцессор позволяет использовать командную строку или определять строки в командном файле (команда MACRO);
- использование «встроенного калькулятора с переменными», которые можно совмещать с командами ввода в командном файле (команда CALC);
- чтение одного командного файла из другого (команда READ);
- возможность гибкого создания и редактирования атомной структуры (команды BOX, FILL, PARTICLE);
- выбор подмножества атомов для различных действий с использованием разнообразных шаблонов (команды SELECT, SET и TAG);
- выполнение действий с выбранными атомами, таких как их перемещение (MOVE), вставка дефектов (SCREW, WAVE), установка типа атома (TYPE), установка массы атома (MASS), вращение выбранных атомов (ROTATE) и т.д.;
- стабилизация заданной температуры вещества (команда CLAMP);
- стабилизация давления, с применением различных алгоритмов «зажима давления» (команды PRESSURE, QUENCH);
- фиксация положения выбранных атомов на линии, в плоскости или в пространстве (команды FIX, CONSTRAIN);
- применение внешней силы к отдельным атомам (команда EXTFORCE);
- применение скоростных коэффициентов демпфирования к выбранным атомам (команда DAMP);
- использование парного потенциала взаимодействия, метода «вложенных» атомов (EAM), потенциала карбида кремния Терсоффа или потенциала Стиллинжера-Вебера (SW) в системе единиц СИ;
- сохранение результатов моделирования для последующего продолжения (команда WRITE STATE);
- сохранение энергетических показателей (команды ESAVE и WRITE ENERGY), атомных координат (команда WRITE PARTICLE) и другой информации в текстовых файлах во время моделирования;
- координаты могут быть сохранены в специальных форматах (команды RCV или COR) для дальнейшего использования с другими программами, входящими в пакет, такими как XMDVIEW (для просмотра графики), WMOVIEC (для просмотра анимации);
- имеются встроенные подпрограммы для представления атомной конфигурации в цвете на оборудовании, использующем язык Postscript (команда PLOT).
Думаю, такой список возможностей XMD вполне удовлетворит даже взыскательного исследователя. Добавим сюда также достаточно подробное руководство пользователя, которое поставляется с пакетом (есть также “on-line” руководство на сайте [1] в разделе Documentation).
Моделирование с помощью МД имеет предел в числе атомов, с которыми работает программа, в значениях между 5 000 и 100 000 атомов. С таким количеством атомов невозможно моделировать реальный объем материала, если только не использовать так называемые «повторяющиеся граничные условия». С повторением граничных условий моделирование проводится в пределах небольшого «кусочка» вещества, который обычно называется «кристаллитом». Атомы, которые проходят через одну границу кристаллита, возвращаются назад через противоположную границу.
Таким образом, с помощью XMD можно проводить различные эксперименты с условиями очень похожими на реальные не отходя от своего любимого персонального компьютера.
Установка программы
Настало время показать читателю, как установить пакет XMD на персональный компьютер. Хотя на сайте проекта [1] в разделе Downloads есть версия программы, портированная под операционную систему (ОС) Windows, разработчиками изначально предполагается для работы XMD использование ОС Unix. Пользователь Windows останется без мультипроцессорной поддержки, использующей стандарт POSIX, а, самое главное, без средств визуализации, написанных с использованием X-Window System и библиотеки Xaw (набора виджетов для реализации простых интерфейсов пользователя), что делает использование клонов Unix, а именно Linux, предпочтительным.
Как показывает опыт, работа с Linux для пользователей персонального компьютера, незнакомых с этой ОС, вызывает некоторые затруднения. Однако тот же опыт говорит, что можно попробовать (или попросить «продвинутого» товарища) установить эту систему в виртуальной машине (например, VirtualBox) на компьютере с Windows и, получить работоспособную среду XMD. В сети Интернет есть подробные описания по установке различных систем на персональный компьютер, в том числе и в виртуальную машину.
Покажем, как установить пакет программ XMD в ОС Linux. Автор использовал для этого Linux Mint 13 (кстати, установка данного дистрибутива, как отмечают многие, даже проще установки Windows).
Для установки XMD (а это придется делать из исходных кодов) на чистый Linux Mint нам понадобится пакет build-essential. Отметим еще раз, что все действия по установке и работе с программой выполняются из командной строки (программа «терминал», xterm или подобная). Итак, запустите терминал и выполните следующую команду:
$ sudo apt-get install build-essential
Введите пароль пользователя и, после установки пакета, скачайте архив XMD с сайта [1] из раздела Downloads. Далее, распаковываем архив и переходим в образовавшийся каталог.
$ cd
$ tar xzvf xmd-2.5.38.tar.gz
$ cd xmd-2.5.38/
Конфигурируем, собираем и устанавливаем программу:$ ./configure
$ make
$ sudo make install
После завершения процедуры make программа XMD (исполняемый файл – xmd записывается маленькими буквами, в среде Unix это важно) будет собрана из исходных кодов и готова к использованию.
Рекомендую сразу установить программу визуализации xmdview. Для этого нам будет необходима библиотека Xaw. В Linux Mint 13 нужные для работы библиотеки файлы устанавливаются с помощью команд
$ sudo apt-get install xutils xutils-dev libxaw7-dev
Перейдя в каталог с исходниками для xmdview$ cd xmd-2.5.38/xmdview-1.1
Выполните сборку$ sh ./Compile
и копируем программу в каталог, который виден переменной PATH$ sudo cp ./xmdview /usr/local/bin
Теперь можно смело приступать к моделированию. Отметим, что вместе с программами и их исходными кодами поставляется множество примеров командных файлов XMD в каталоге examples. Их изучение поможет быстро освоить возможности пакета.
Первые шаги в XMD
Для ознакомления с работой XMD можно порекомендовать построить кристаллическую решетку какого-либо вещества и просмотреть ее с помощью программы визуализации пакета - xmdview. Подобные картинки с успехом может использовать в своей работе преподаватель химии, физики и т.п. В документации xmd на сайте проекта есть пример построения кристаллической решетки алмаза. Алмаз (бриллиант) – сам по себе является «королем» драгоценных камней, но построить и посмотреть его кристаллическую решетку в XMD очень просто.
Для этого необходимо с помощью обычного текстового редактора (например, того же gedit – одной из «стандартных» программ Linux Mint) создать текстовый файл, содержимое которого показано в Листинге 1.
Листинг 1. Содержимое файла diamond.txt
box 5/4 5/4 5/4
fill particle 2
1 0 0 0
1 1/4 1/4 1/4
fill cell
0 1/2 1/2
1/2 0 1/2
1/2 1/2 0
fill go
scale 3.57
write cor diamond.cor
fill particle 2
1 0 0 0
1 1/4 1/4 1/4
fill cell
0 1/2 1/2
1/2 0 1/2
1/2 1/2 0
fill go
scale 3.57
write cor diamond.cor
Сохраним файл как diamond.txt. Некоторые пояснения к командам, использованным в этом файле. В кристаллографии, знание основ которой однозначно пригодится для работы с любыми программами молекулярной динамики, широко используется понятие “элементарная ячейка”. Фактически это минимальный воображаемый объем кристалла, параллельные переносы (трансляции) которого в трех измерениях позволяют построить трехмерную кристаллическую решетку в целом (см. Википедия, “элементарная ячейка”).
Установлено, что элементарную ячейку алмаза можно построить с помощью трансляции координат всего двух атомов углерода в направлениях с относительными координатами по осям x, y, z трехмерного пространства:
0 1/2 1/2
1/2 0 1/2
1/2 1/2 0
1/2 0 1/2
1/2 1/2 0
что и записано в diamond.txt с помощью команд box, fill particle, fill cell и fill go. Отметим, что box задает размер кристаллита количеством повторяющихся элементарных ячеек. Размер же элементарной ячейки в ангстремах (10-10 м) определяется с помощью команды scale.
Следующая конструкция “write cor diamond.cor” определяет имя файла, в который будут записаны координаты атомов кристаллической решетки алмаза.
После того как файл diamond.txt сохранен на диске. Перейдите в каталог, где лежит этот файл и запустите XMD (с именем командного файла вместо параметра) в терминале:
$ cd nuzhny_katalog
$ xmd diamond.txt
Итогом выполнения расчета XMD будет файл diamond.cor с координатами атомов кристаллической решетки алмаза, посмотреть который можно с помощью программы xmdview (не пытайтесь просмотреть такой файл с помощью текстового редактора, т.к. он имеет специальный двоичный формат):
$ xmdview diamond.cor
Вот что получается в результате выполнения этой команды (см. рис.1).
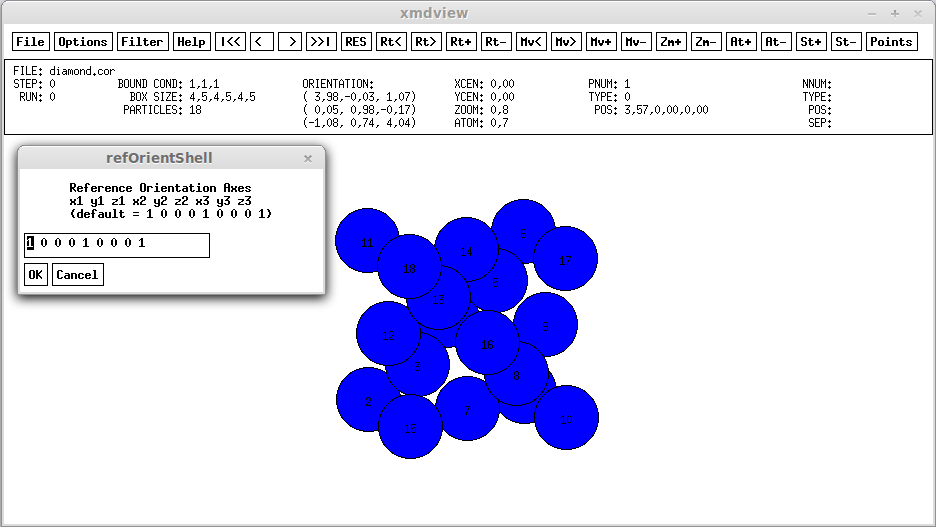
Рис.1 Программа xmdview — структура элементарной ячейки алмаза (файл diamond.cor)
Используя интерфейс xmdview можно, например, покрутить трехмерное изображение (кнопки "Rt>", "Rt+", "Rt-"), посмотреть где находятся центры атомов или переключиться на просмотр частиц в виде сфер (кнопка "Points/Circles"), уменьшить/увеличить масштаб ("Zm+", "Zm-") или размеры частиц ("At+", "At-"), посмотреть координаты конкретной частицы и т.д. Интерфейс программы несложный, легко изучается «методом тыка», хотя есть и помощь, правда на английском.

рис.2 «Нанотрубка» - пример из samples/nanotube2
Несколько слов о межатомных потенциалах
Следующим этапом в изучении XMD, безусловно, является построение «динамических» симуляций. И здесь, конечно, не обойтись без «межатомных потенциалов» или, как их еще называют в отечественных источниках, «потенциалов парного взаимодействия» (ППВ).
Физический смысл их применения в МД достаточно прост и не выходит за рамки классической физики. Потенциальная энергия взаимодействия двух атомов записывается как функция расстояния между ними. Атомы в решетках при определенных условиях могут сталкиваться и их столкновения очень похожи на столкновения упругих шаров. Таким образом, общая энергия системы атомов складывается из потенциальной энергии взаимодействия всех частиц между собой и суммы кинетических энергий атомов.
XMD может использовать также так называемый «метод вложенных атомов» (EAM - embedded atom method), где при учете потенциальной энергии системы, кроме суммы энергий взаимодействия атомов между собой, еще добавляется энергия взаимодействия частицы с электронным облаком соседей по кристаллической решетке. Атом при этом считается «вложенным» в некое подобие «электронной плотности».
Все функции взаимодействия записываются в виде рядов значений по определенным правилам в отдельный файл, который называется «файлом потенциалов». Такой файл добавляется в командный с помощью конструкции READ.
В пакете XMD есть примеры (см. папку examples) использования файлов потенциалов железа, сплава Ni-Al и т.д. Также файлы потенциалов для других веществ и соединений можно найти на сайте проекта [1] в разделе «EAM potentials». (О применении межатомных потенциалов в xmd читайте в следующей статье.)
***
Итак, в статье мы познакомились с одним из способов компьютерного моделирования - методом молекулярной динамики, а также с одной из программ, реализующей данный метод на персональном компьютере – XMD. Подробно был рассмотрен процесс установки программы на компьютер с ОС Linux Mint. Была также попытка «заглянуть» внутрь кристаллической решетки алмаза. Вы еще не почувствовали себя «творцом»? Тогда изучайте программы молекулярной динамики дальше и обязательно это почувствуете! Успехов!
Ссылки:
1. Сайт проекта XMD - http://xmd.sourceforge.net/
2. Молекулярная динамика пептидов, К.В. Шайтан - http://old.bioeng.ru/doc/stat/moldyn.htm
3. Википедиа Метод классической молекулярной динамики - https://ru.wikipedia.org/wiki/%D0%9C%D0%B5%D1%82%D0%BE%D0%B4_%D0%BA%D0%B...
4. Молекулярная динамика на персональном компьютере. Учебное пособие. Пестряев Е.М. - http://www.rusoil.net/pages/14246/Pestryaev_%20Molek.dinamika.pdf
5. Методы молекулярной динамики для моделирования физических и биологических процессов. Х.Т. Холмуродов и др. - http://www1.jinr.ru/Pepan/v-34-2/v-34-2-5.pdf
6. Программа метода молекулярной динамики, ориентированная на моделирование физических процессов в высокотемпературных полупроводниках. В.М.Дядин, В.Г.Чудинов, Л.И.Гондырева, В.А.Бычков, АН СССР, Уральское отделение, Физико-технический институт №1537-В 91, Ижевск 1991г.
7. Моделирование теплофизических свойств веществ методами молекулярной динамики с использованием параллельных вычислений
- http://www.dissercat.com/content/modelirovanie-teplofizicheskikh-svoistv...
- Войдите или зарегистрируйтесь, чтобы отправлять комментарии
|
Позиция сайта в рейтинге:
|
Дружественные сайты: |